Biofizikalni svet modeliranja

Poslušate prvo letošnjo jesensko oddajo Frequenza della Scienza. Danes se bomo spogledovali s fizikalno teorijo ter kemijskimi in biološkimi eksperimenti, vendar bomo ostali v varnem zavetju računalnika in numeričnih simulacij. Govorili bomo o numeričnih pristopih, ki se uporabljajo v interdisciplinarni vedi biofiziki.
Biofizika se ukvarja z zelo širokim spektrom problemov. Za razumevanje bioloških procesov uporablja fizikalne metode in pristope. Spopada se s problemi iz biologije, medicine in farmacije ter jih skuša razložiti s fizikalnimi teorijami, raznimi eksperimenti in numeričnimi simulacijami. Več o tem v nadaljevanju, najprej pa si poglejmo, zakaj moramo uporabljati numerične modele.
Za trenutek se vrnimo k osnovnošolski fiziki. Z Newtonovimi zakoni lahko opišemo gibanje Zemlje okoli Sonca. V takem poenostavljenem modelu znamo natančno določiti njuno pozicijo v vsakem časovnem koraku. »Poenostavljenem« pravimo zato, ker takoj, ko jima dodamo tretje telo, denimo Mars, naletimo na zaplete. Sistem postane nestabilen za večino začetnih pogojev in težko uganemo stabilne lege teles. Seveda pa je teh v našem osončju veliko več. Enako velja tudi za ostale fizikalne sisteme, v katerih opazujemo interakcije med delci. Kaj hitro naletimo na težave, ko moramo opisati gibanja kvadrilijona teles, kolikor približno znaša Avogadrovo število. S tem se zna dobro spoprijeti statistična fizika, saj znamo z razumevanjem obnašanja in gibanja gradnikov snovi napovedati njene lastnosti. A več o tem v nadaljevanju.
Uporabljamo numerične metode. Z raznimi algoritmi naredimo približek analitičnega modela in ga tako poenostavljenega rešimo. Pri reševanju Newtonovih enačb v enostavnem sončnem sistemu Sonca, Zemlje in Lune to pomeni, da odvode aproksimiramo s povprečenjem hitrosti v zaporednih časovnih korakih. Tako dobimo približek rešitve v vsakem časovnem koraku in za ves sistem lahko napovemo, kako se bo obnašal v katerem koli trenutku.
Pri biofizikalnih problemih pa so naša opazovana telesa veliko manjša kot v astronomiji. Nadaljuje doktorica Julija Zavadlav z Univerze v Münchnu.
Kako pa numerične simulacije dopolnijo teoretične modele in eksperimente, razloži Matej Praprotnik s Kemijskega inštituta v Ljubljani.
S teoretičnim razumevanjem problema si lahko zastavimo sistem, ki ga potem simuliramo.
Vzemimo si trenutek in premislimo, kar smo ravnokar slišali. Pri numeričnih simulacijah želimo mikroskopske lastnosti sistema povezati z makroskopskimi. V najpreprostejšem primeru opazujemo škatlo z delci zraka. V mikroskopski sliki opazujemo delce, kako se gibljejo in kako trkajo ali interagirajo med sabo. To lahko numerično modeliramo s poenostavljenimi modeli, kot rešitev pa dobimo njegove makroskopske lastnosti. Če gre za zrak v škatli, dobimo denimo njegovo gostoto, tlak, volumen in podobno.
Kot smo slišali, za poenostavitev modela uporabljamo veliko trikov. Da nam ni treba obravnavati ogromnega števila delcev, si zamislimo periodične robne pogoje na mejah našega sistema. Še en trik uporabimo pri elektrostatskih interakcijah. Če so delci, ki jih opazujemo, nabiti, moramo pri reševanju Newtonovih enačb gibanja upoštevati tudi coulombsko [kulOmsko] silo. Ta interakcija je dolgosežna, kar bi numerično obravnavo zelo otežilo, saj bi morali opazovati interakcijo med vsemi delci sistema, tudi tistimi najbolj oddaljenimi. Reši nas pojav senčenja coulombskega potenciala, zaradi katerega pade sila na nič že pri nekaj nanometrih.
Ste na valovih Radia Študent v oddaji Frequenza della Scienza. Tema današnje oddaje so numerični pristopi k biofizikalnim sistemom.
Nazadnje smo spoznali poenostavitve, ki jih lahko pri obravnavi privzamemo, ko želimo reševati Newtonove enačbe gibanja za ogromne sisteme delcev. Kot pa smo že slišali od sogovorcev, je obravnava vsakega delca posebej računsko zelo potratna. Zdaj bomo podrobneje spoznali dva modela, ki se uporabljata v biofizikalnih simulacijah in na katera smo ves čas namigovali.
Začnimo pri metodi Monte Carlo. Najpreprosteje jo je opisati z izračunom vrednosti števila pi. Igramo pikado s kvadratno tarčo, ki ji vrišemo krog. V tarčo mečemo puščice in si zapisujemo, koliko jih vržemo in koliko jih zadene krožni del tarče. Ob predpostavki, da smo z vsemi puščicami zadeli tarčo, nam razmerje med meti, ki so pristali v krogu, in vsemi meti poda razmerje med ploščinama kroga in kvadrata. Ta je ravno pi četrtin.
Pri vseh implementacijah metode Monte Carlo sledimo enakemu postopku. Najprej definiramo vse možne izide, ki se lahko zgodijo v enem koraku. V našem primeru je to zadetek katerega koli dela tarče. V naslednjem koraku izvedemo naključni žreb med možnimi izidi oziroma vržemo puščico. Preverimo, ali je to, kar smo naredili v prejšnjem koraku, za nas ugodno ali ne. Pri izračunu števila pi smo pogledali, ali smo zadeli v okrogli del tarče ali ne. V zadnjem koraku pa zberemo vse rezultate. S povečevanjem števila metov v tarčo se tako vedno bolj bližamo vrednosti števila pi.
Zavladav pojasni, kako se metoda uporablja v biofizikalnih simulacijah.
Taka implementacija metode Monte Carlo nam da končne konfiguracije sistema, ničesar pa ne more povedati o njegovi dinamiki. Doktorica Zavadlav zato opiše še drugo uporabno metodo, metodo molekulske dinamike.
Molekulska dinamika tako ni nič drugega kot problem več teles – reševanje enačb gibanja za ogromno število delcev. Tako pri vsakem časovnem koraku dobimo informacijo o hitrosti in koordinatah delcev. Tako lahko denimo modeliramo difuzijo delcev v sistemu in, kot je bilo omenjeno v izjavi, izračunamo hitrosti zvijanja proteinov. Seveda je to zelo poenostavljen pogled na metodo, pri katerem predpostavljamo, da so telesa sistema kroglice. V kompleksnejših sistemih moramo upoštevati kvantnomehanske sile, ki so računsko bistveno potratnejše. Več o tem v nadaljevanju.
Magic Panda — Life is Elsewhere
Še vedno ste na frekvenci 89,3 MHz in poslušate oddajo znanstvene redakcije Frequenza della Scienza o računalniških simulacijah v biofiziki. Govorili smo že o velikih računalniških potrebah, vendar še nismo pojasnili, kakšne sploh so. Poskusimo to na grobo oceniti.
Za opis dinamike dveh prostih delcev moramo hraniti podatke o njunih hitrostih in koordinatah v vsakem koraku. V treh dimenzijah moramo tako v vsakem koraku shraniti šest novih podatkov, starih pa ne smemo zavreči, saj jih bomo potrebovali. Pri večanju števila delcev se tako časovna zahtevnost linearno povečuje.
Z upoštevanjem interakcij med delci časovno zahtevnost še povečamo. V vsakem koraku moramo namreč izračunati interakcijo z vsemi ostalimi delci, skupaj torej število delcev na kvadrat. Časovna zahtevnost se tako poveča že na kvadratično. Za sto delcev v izbranem sistemu moramo tako – zelo približno – izračunati 10.000 interakcij v vsakem časovnem koraku. Pri poenostavljenih bioloških sistemih želimo simulirati milijon delcev, kar zahteva od računalnika bilijon operacij v vsakem koraku.
Prenosni računalnik avtorice oddaje deluje s frekvenco 3 GHz, kar pomeni, da lahko v vsaki sekundi naredi milijardo operacij. Tako bi za izračun bilijona operacij potreboval 1000 sekund ali okoli 17 minut. A tu gre le za en časovni korak, mi pa želimo napovedati dinamiko za daljše časovno obdobje. Nevedne še opozorimo, da z večanjem časovnega koraka izgubljamo natančnost modela. Tako ne moremo v enem koraku preskočiti kar sto let procesov.
O računski zahtevnosti nadaljuje doktor Praprotnik.
Superračunalnik v Mariboru, ki lahko računa petaflope oziroma bilijardo operacij na sekundo, bi za tistih bilijon operacij, ki ga je navaden prenosni računalnik opravil v 17 minutah, porabil tisočinko sekunde, kar je milijonkrat manj.
Kako pa je z računsko zahtevnostjo predstavljenih simulacij Monte Carlo in molekulske dinamike, pojasni Zavadlav.
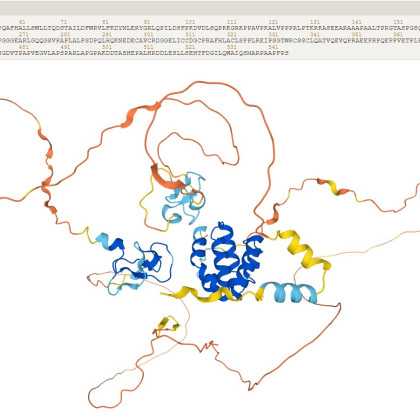
Današnja sogovornica je že omenila, da lahko za napoved zvijanja proteinov uporabimo tako molekulsko dinamiko kot tudi simulacijo Monte Carlo. Na tem področju je sicer veliko uspešnejša nevronska mreža v osredju programa AlphaFold, o katerem smo v znanstveni redakciji že poročali.
Tak pristop je seveda veliko cenejši od eksperimentalnih. Ne potrebujemo dolgotrajne priprave vzorcev, pri čemer obstaja tudi možnost, da priprava niti ne bo uspela. Ne potrebujemo niti dragih sinhrotronov, NMR-spektroskopov ali elektronskih mikroskopov. Potrebujemo le dovolj zmogljive računalnike, da lahko poganjajo naše simulacije.

Oddajo je pripravila Tina.
Brala Rasto in Muri.
Tehniciral Filip.








Dodaj komentar
Komentiraj