AlphaFold2 – od sekvence k strukturi
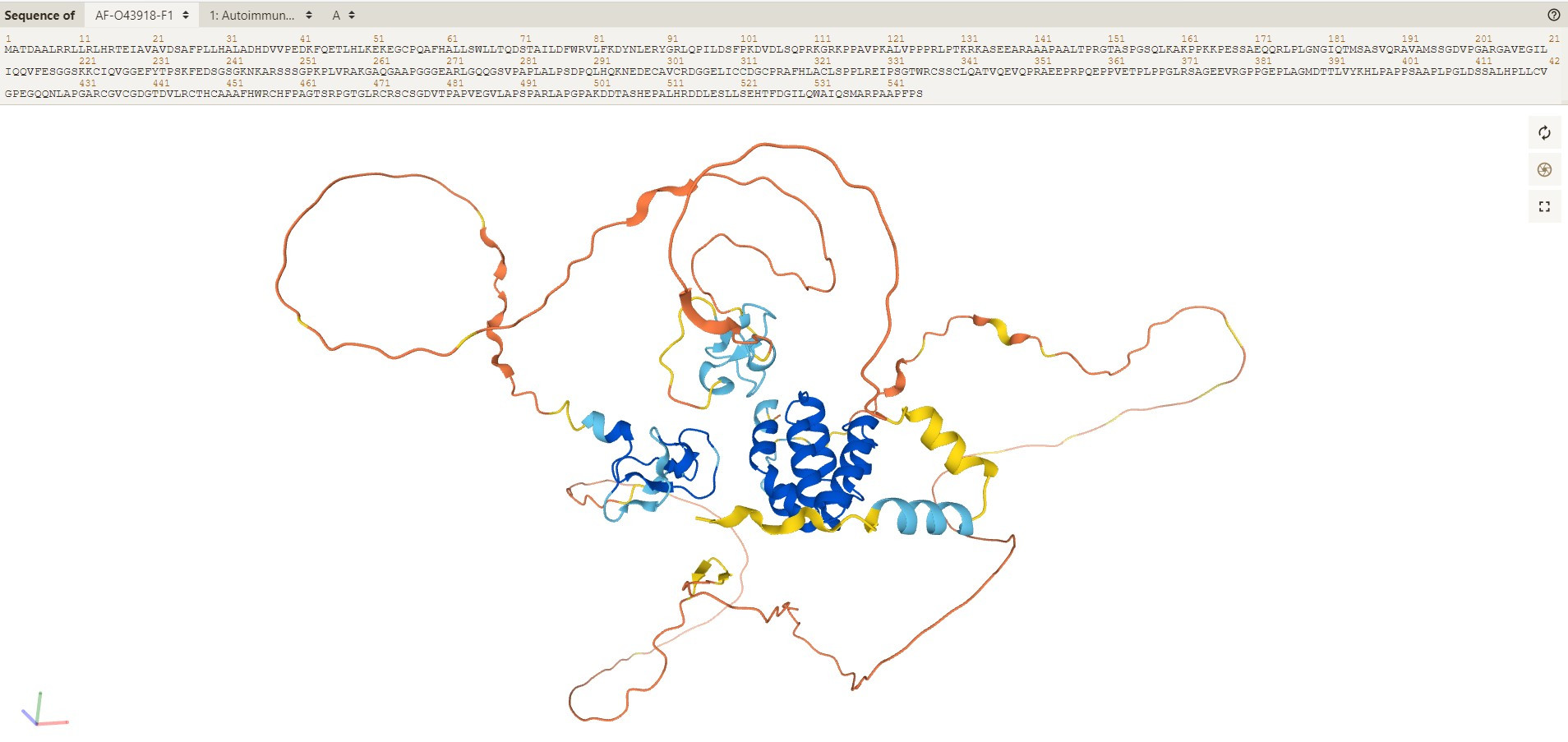
V tokratnem Znanstvenem komentarju se posvečamo verjetno enemu od pomembnejših dosežkov moderne biologije. Potem ko je lansko jesen računalniški program za napoved tridimenzionalnih struktur proteinov AlphaFold2 gladko pometel s konkurenco in dosegel rezultat, ki je v večini primerov povsem primerljiv z eksperimentalno določenimi strukturami, so razvijalci programa nedavno objavili še celotno arhitekturo in izvorno kodo programa. Še več, v sodelovanju z Evropskim bioinformatskim inštitutom so ustvarili javno dostopno zbirko strukturnih modelov proteinov celotnega človeškega proteoma in več dvajset drugih modelnih organizmov.
Najprej nekaj ozadja. Proteini so gotovo najpomembnejše molekule življenja. Tako ali drugače so udeleženi v čisto slehernem procesu v živi celici. Navodila za njihovo izgradnjo so shranjena v genomu v obliki molekule DNA. Molekularna koda v obliki štirih črk, ki predstavljajo štiri nukleotide, se na ribosomu prevede v polipeptidno verigo, ki jo sestavlja dvajset aminokislinskih ostankov. Interakcije aminokislinskih ostankov s topilom in drugimi aminokislinskimi ostanki v nastali verigi poskrbijo, da nastala polipeptidna veriga v celici zavzame edinstveno tridimenzionalno strukturo. Prav ta prostorska razporeditev aminokislinskih ostankov v nativni strukturi proteina je tista, ki proteinu omogoča izvršitev njegove biološke funkcije, pa najsi gre za encimatsko pospešitev določene kemijske reakcije, prenos signala znotraj celice ali pa izgradnjo citoskeleta.
Poznavanje tridimenzionalne zgradbe proteina je tako ključno za razumevanje biološke funkcije. A določitev strukture je eksperimentalno precej težaven proces. O tem priča tudi realtivno majhno število rešenih struktur proteinov, ki jih raziskovalci in raziskovalke shranjujejo v podatkovni zbirki Protein Data Bank. Danes tako tam najdemo okoli 180 tisoč rešenih struktur, skoraj 160 tisoč od njih proteinskih. Kljub temu da se navedeno število morda zdi veliko, predstavlja v resnici zgolj majhno kapljico v morju. Denimo že za človeški proteom, ki zapisuje okoli dvajset tisoč proteinov, poznamo strukture zgolj okoli tretjine vseh človeških proteinov, in še teh ne vedno v celoti.
Zaradi težav, povezanih z eksperimentalno določitvijo, se raziskovalci in raziskovalke že desetletja trudijo razvozlati povezavo med primarnim aminokislinskim zaporedjem, torej sekvenco črk, ki predstavljajo aminokislinske ostanke, in tridimenzionalno strukturo proteina. V ta namen poskušajo razviti računalniški program, ki bi danim aminokislinskim zaporedjem napovedal pripadajočo tridimenzionalno strukturo. Organizirali so pravo tekmovanje med razvijalci takšnih programov, ki poteka na vsaki dve leti že od leta 1994. Na tekmovanju, imenovananem Critical Assessment of Structure Prediction, krajše CASP, dobi vsaka ekipa nalogo čimbolj natančno napovedati strukture proteinov, za katere so jim podana zgolj njihova aminokislinska zaporedja. Strukture teh proteinov so sicer poznane ozkemu krogu raziskovalcev, ki so jih že uspešno rešili, a jih še niso deponirali v javno dostopno bazo struktur. Zmago doseže ekipa, katere računalniški program se kar najbolj približa rezultatom eksperimentalno določenih struktur.
AlphaFold2, računalniški program za napoved tridimenzionalne strukture danega aminokislinskega zaporedja, je razvilo britansko podjetje DeepMind, ki deluje v skupini podjetij Alphabet in je tako pravzaprav hčerinsko podjetje spletnega velikana Googla. DeepMind je v preteklosti že zaslovel s svojim šahovskim programom, še bolj pa s programom AlphaGo, ki je kot prvi računalniški program uspel premagati najboljše igralce strateške igre Go. Podjetje se osredotoča predvsem na razvoj in implementacijo novih tehnologij umetne inteligence za reševanje težjih računskih problemov. Z AlphaFold so se razvijalci in razvijalke podjetja DeepMind od namiznih iger preusmerili k enemu od večjih problemov molekularne biologije, torej napovedi tridimenzionalne strukture proteina iz aminokislinskega zaporedja. Rešitve, ki jih je prikazal AlphaFold2, se v večini primerov le malo razlikujejo od eksperimentalne določitve z visokoresolucijskimi tehnikami X-žarkovne kristalografije ali nuklearne magnetne resonance.
Delovanje programa kakopak temelji na orodjih umetne inteligence, to je nevronskih mrežah, in na metodi globokega učenja. Poudariti sicer velja, da AlphaFold nikakor ni prvi ali edini program na področju napovedovanja struktur proteinov, ki temelji na principu strojnega učenja. Nevronska mreža, ki je osrednja v algoritmu AlphaFold2, je bila natrenirana na javno dostopnih podatkih iz arhiva struktur na Protein Data Bank. Poleg tega pa pri izgradnji modela strukture program upošteva še evolucijske podatke, ki tudi nosijo informacijo o prostorski soodvisnosti aminokislinskih ostankov v zaporedju oziroma tridimenzionalni strukturi. Uspeh AlphaFold2 se, kot pojasnjujejo strokovnjaki s področja umetne inteligence, skriva v uporabi modernih pristopov in številnih inovativnih inženirskih rešitvah razvijalcev DeepMind. Ena od tehnik, ki naj bi bistveno izboljšala napovedno moč programa, je tako imenovana pozornost, angleško »attention«, ki predstavlja tehniko, ki omogoči nevronski mreži osredotočenje na pomembnejše dele vhodnih informacij, denimo na dele aminokislinskega zaporedja, ki nosijo več informacij za tridimenzionalno strukturo proteina.
Nazaj k izjemnemu rezultatu, ki ga je dosegel AlphaFold2. Kaj nam in širši znanstveni skupnosti tako dober rezultat sploh pomeni in česa ne? Kljub številnim pomislekom akademske skupnosti, da bo DeepMind držal obljubo in objavil izvorno kodo programa, smo v mesecu juliju poleg te v sodelovanju z Evropskim bioinformatskim inštitutom dobili še podatkovno bazo napovedi struktur proteinov celotnega človeškega proteoma in več dvajset drugih modelnih organizmov. Takšna nabirka zelo dobrih napovedi tridimenzionalnih struktur proteinov bo omogočila vrsto novih aplikativnih, pa najbrž tudi bolj bazičnih spoznanj. Modeli struktur proteinov, katerih tridimenzionlana zgradba še ni bila znana in so udeleženi v bolezenskih procesih, bodo denimo omogočili bistveno hitrejši in racionalnejši razvoj novih potencialnih učinkovin. S pomočjo strukturnih modelov pa bomo sedaj lahko še bolje razumeli, kako sta struktura in funkcija proteina zares povezani.
Morda se bo kdo vprašal, kaj AlphaFold2 pomeni za strukturno biologijo. Ali lahko že kar zapremo sinhrotrone, izključimo elektronske mikroskope in magnete? Odgovor je seveda jasen in gotov ne. AlphaFold2 tudi za eksperimentalno strukturno biologijo pomeni pomembno pridobitev, s katero se bodo lahko raziskovalci lotevali težjih problemov, kot so denimo strukturne študije kompleksov proteinov z nukleinskimi kislinami ali pa strukturno kompleksnejši makromolekularni sistemi, ki jih danes pogosto raziskujemo z uporabo krioelektronske mikroskopije. Predstavljeni program bo pomembno vplival tudi na še boljše zavedanje vloge in pomena dinamike proteinov. Pričakujemo lahko še več študij, usmerjenih k dinamičnim lastnostim proteinske strukture.
Ozrimo se na koncu še na pomembne omejitve, ki jih AlphaFold2 kljub svoji veliki napovedni moči vseeno ima. Znova velja poudariti, da lahko veliko zaslug za uspeh algoritma pripišemo dolgi zgodovini določanja struktur bioloških makromolekul in podatkovni bazi Protein Data Bank, saj so se nevronske mreže učile prav iz teh podatkov. To pa prinaša tudi svojevrstno omejitev, saj, kot smo že omenili, trenutni nabor struktur v javnem arhivu predstavlja le en, pravzaprav precej majhen del možnega sekvenčnega prostora. To bi lahko vodilo v delno pristranskost nevronske mreže, ki bi se lahko izrazila predvsem pri novih, evolucijsko oddaljenih, nepoznanih zaporedjih. Poseben problem bi tako lahko predstavljala predvsem umetna, de novo aminokislinska zaporedja, pri katerih algoritem ne uspe pridobiti kvalitetne evolucijske informacije s poravnavo homolognih aminokislinskih zaporedij.
Dodaten izziv AlphaFold2 predstavljajo zaporedja ali deli njih, ki jasne tridimenzionalne strukture sploh ne zavzamejo.
AlphaFold2 bo nedvomno dodobra transformiral področje strukturne biologije in še pospešil razvoj novih podobnih računalniških orodij, ki bodo prispevala k boljšemu razumevanju življenjskih procesov in tudi patologij na molekularni ravni. Pomembnost razkritja programske kode in lansiranje podatkovne baze strukturnih modelov proteinov številnih proteomov zato lahko postavimo celo ob bok razkritju prvega osnutka človeškega genoma izpred dvajsetih let.

Zaključimo z nekoliko bolj fundamentalnim aspektom AlphaFold2 in se vprašajmo, ali smo zdaj končno in dokončno odgovorili na tako imenovani problem zvijanja proteinov, angleško protein folding problem, ki ga je že v šestdesetih letih z znamenitimi eksperimenti na ribonukleazi formuliral Christian Anfinsen. Za potrebo razmisleka privzamemo, da kljub nekaterim pomembnim omejitvam, ki smo jih predstavili, AlphaFold2 dano aminokislinsko zaporedje zna skoraj vedno prevesti v prostorsko porazdelitev koordinat atomov proteina. To sicer res predstavlja pomemben korak k razrešitvi protein folding problema, a pojavi se pomembna dilema. V resnici namreč sploh ne vemo, zakaj je algoritem takšno strukturo napovedal. Tako nam umanjka ključen, podroben biofizikalni uvid, ki bi odgovoril na vprašanje, zakaj protein zavzame prav takšno strukturo, kot jo je program tudi napovedal. AlphaFold2 je tako zaenkrat zgolj izjemno uporabna črna škatla. Ob razvoju naprednih računalniških algoritmov za napoved strukture proteinov se tako poraja pomembno vprašanje, ki v resnici sega še daleč onkraj meja strukturne biologije: ali bomo lahko iz zmogljivih, kompleksnih računalniških algoritmov kdaj pridobili globlji biofizikalni uvid ali bodo ti programi služili zgolj kot izjemno zmogljive črne škatle.










Dodaj komentar
Komentiraj